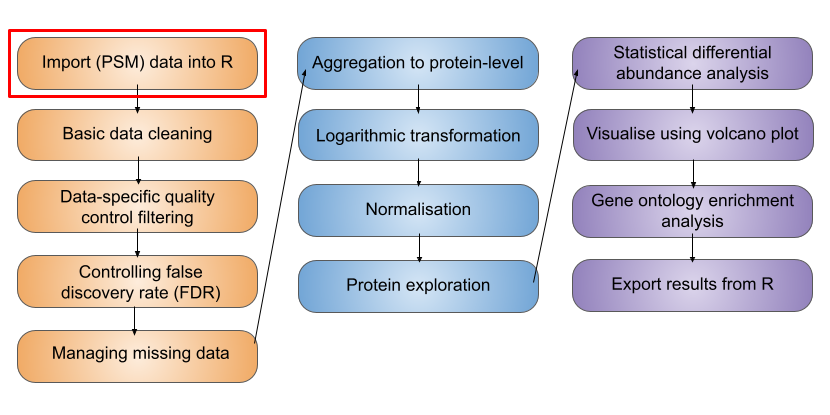
2 Import and infrastructure
TipLearning Objectives
- Understand the structure of
SummarizedExperimentandQFeaturesobjects and how the two are related - Know how to import data into a
QFeaturesobject - Demonstrate how to access information from each part of a
QFeaturesobject by indexing and usingassay,rowDataandcolData
2.1 The data structure
MS-based quantitative proteomics data is typically represented as a matrix in which the rows represent features (PSMs, peptides or proteins) and the columns contain information about these features, including their quantification measurements across several samples. As we saw before, this type of quantitative proteomics matrix can be generated by using third party softwares to process raw MS data. Moreover, the software usually outputs a quantitative matrix for each data level (e.g., PSM, peptide, peptide groups, protein groups). In this lesson we will explain how we can make use of R/Bioconductor packages with dedicated functions and data structures to store and manipulate quantitative proteomics data.
We begin with exporting our data at the lowest data-level. In this DDA TMT experiment this is the PSM level. This .txt file contains for every PSM identified the relative abundance of each PSMs across each sample as well as columns with PSM meta-data.
We will be using the Quantitative features for mass spectrometry, or QFeatures, Bioconductor package to import, store, and manipulate our data. Before we import the data into R using QFeatures it is first necessary to understand the structure of a QFeatures object and SummarizedExperiment object.
2.2 The structure of a SummarizedExperiment
To simplify the storage of quantitative proteomics data we can store the data as a SummarizedExperiment object (as shown in Figure 2.1).SummarizedExperiment objects can be conceptualised as data containers made up of three different parts:
The
assay- a matrix which contains the quantitative data from a proteomics experiment. Each row represents a feature (a PSM, peptide or protein) and each column contains the quantitative data (measurements) from one experimental sample.The
rowData- a table (data frame) which contains all remaining information derived from an identification search (i.e. every column from your identification search output that was not a quantification column). Rows represent features but columns inform about different attributes of the feature (e.g., its sequence, name, modifications).The
colData- a table (data frame) to store sample metadata that would not appear in the output of your identification search. This could be for example, the cell line used, which condition, which replicate each sample corresponds to etc. Again, this information is stored in a matrix-like data structure.
Finally, there is also an additional container called metadata which is a place for users to store experimental metadata. For example, this could be which instrument the samples were run on, the operators name, the date the samples were run and so forth. We will focus on populating and understanding the above 3 main containers within a SummarizedExperiment.
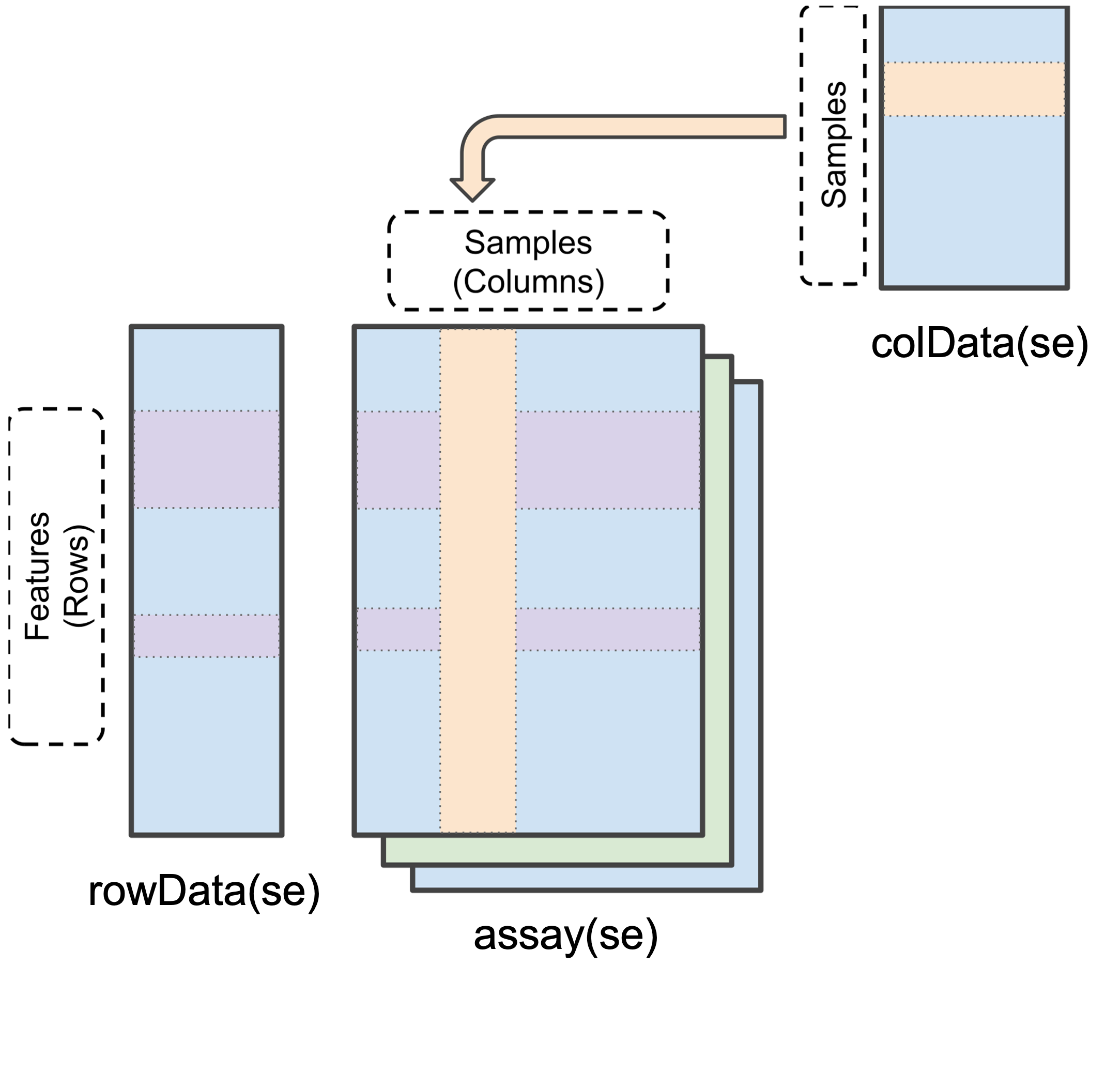
SummarizedExperiment object in R (modified from the SummarizedExperiment package)
Data stored in these three main areas can be easily accessed using the assay(), rowData() and colData() functions, as we will see later.
2.3 The structure of a QFeatures object
Whilst a SummarizedExperiment is able to neatly store quantitative proteomics data at a single data level (i.e., PSM, peptide or protein), a typical data analysis workflow requires us to look across multiple levels. For example, it is common to start an analysis with a lower data level and then aggregate upward towards a final protein-level dataset. Doing this allows for greater flexibility and understanding of the processes required to clean and aggregate the data.
A QFeatures object is essentially a list of SummarizedExperiment objects. However, the main benefit of using a QFeatures object over storing each data level as an independent SummarizedExperiment is that the QFeatures infrastructure maintains explicit links between the SummarizedExperiments that it stores. This allows for maximum traceability when processing data across multiple levels e.g., tracking which PSMs contribute to each peptide and which peptides contribute to a protein (Figure 2.2, modified from the QFeatures vignette with permission).
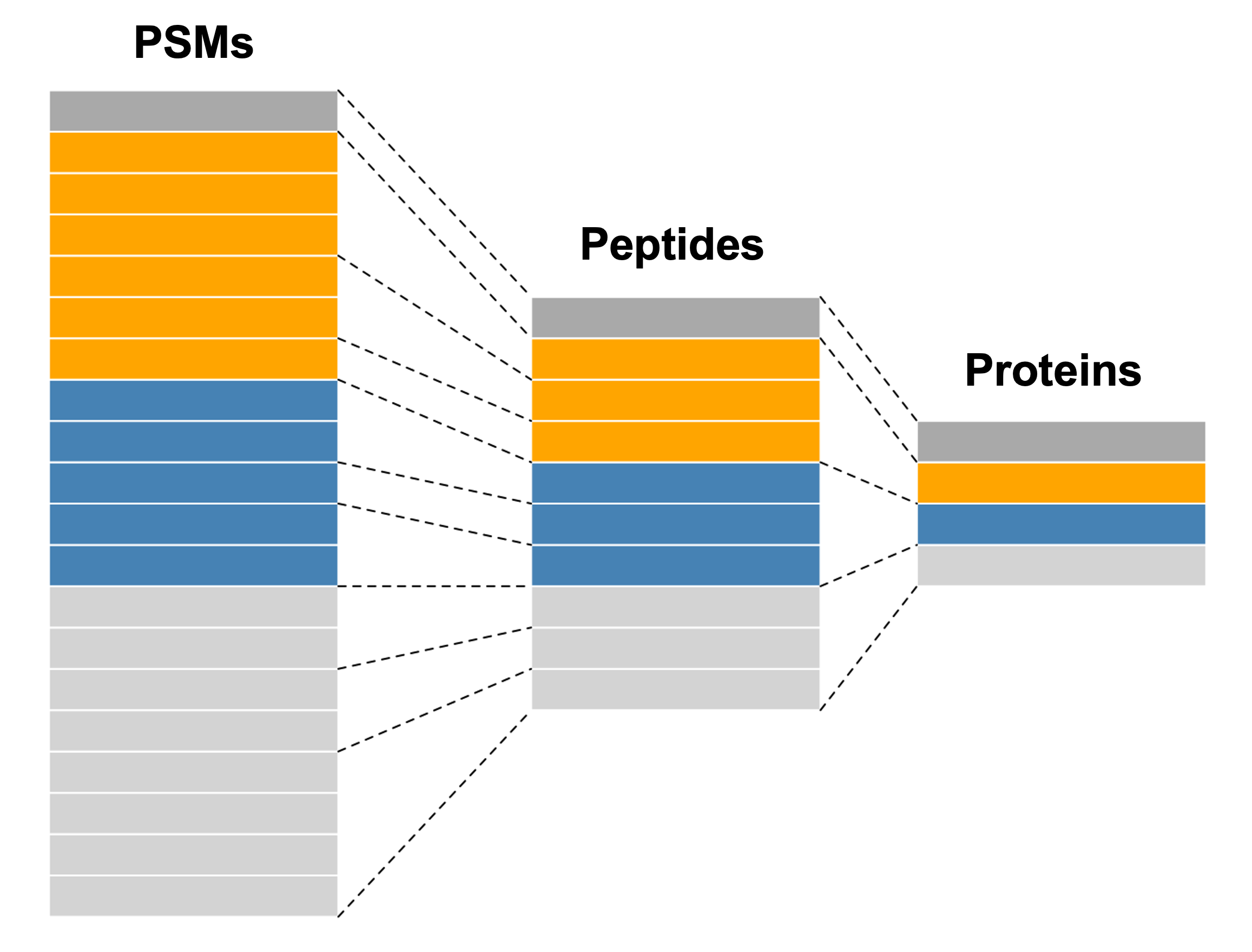
QFeatures object.
NoteQFeatures nomenclature
When talking about a QFeatures object, each dataset (individual SummarizedExperiment) can be referred to as an set (short for dataset). Previously, each dataset was referred to as an experimental assay, and some older resources may still use this term. However, the experimental assay should not be confused with the quantitative matrix section of a SummarizedExperiment, which is called the assay data.
In order to generate the explicit links between data levels, we need to import the lowest desired data level into a QFeatures object and aggregate upwards within the QFeatures infrastructure using the aggregateFeatures function, as we will later see in this course. If two SummarizedExperiments are generated separately and then added into the same QFeatures object, there will not automatically be links between them. In this case, if links are required, we can manually add links using the addAssayLink() function.
The best way to get our head around the QFeatures infrastructure is to import our data into a QFeatures object and start exploring.
2.4 Packages and working directory
During this course we will use several R/Bioconductor packages.
Let’s begin by opening RStudio and loading these packages into our working environment.
library("QFeatures")
library("limma")
library("factoextra")
library("org.Hs.eg.db")
library("clusterProfiler")
library("enrichplot")
library("patchwork")
library("tidyverse")
library("pheatmap")
library("ggupset")
library("here")Set your working directory to Course_Materials where you will find all the relevant data and R code files for this course. This can be achieved using the setwd function or by going to the menu Session -> Set Working Directory -> Choose Directory, in RStudio. Type list.files into your R console to check you can see the materials.
2.5 Importing data into R
There are several ways in which data can be imported into a QFeatures object. The most straightforward way is to first read the data into R as a data.frame using the read.delim function, and then import this data.frame into a QFeatures object.
Arguments to pass to readQFeatures:
assayData= Adata.frame, or any object that can be coerced into adata.frame, holding the dataquantCols= an index for columns containing quantitative data. Can be a numeric index of the columns or a character vector containing the column namesname= name we want to give our firstexperiment, in theQFeaturesobject
Let’s read in the total proteome dataset from our experiment which is has been output from Proteome Discoverer at the PSM level. The file is called cell_cycle_total_proteome_analysis_PSMs.txt.
The first step is to read the data into R as a data.frame. We use the read.delim function.
df <- read.delim("data/cell_cycle_total_proteome_analysis_PSMs.txt")Before we can create a QFeatures object we need to first identify which columns the quantitation data is stored in.
df %>% names() [1] "Checked" "Tags"
[3] "Confidence" "Identifying.Node.Type"
[5] "Identifying.Node" "Search.ID"
[7] "Identifying.Node.No" "PSM.Ambiguity"
[9] "Sequence" "Annotated.Sequence"
[11] "Modifications" "Number.of.Proteins"
[13] "Master.Protein.Accessions" "Master.Protein.Descriptions"
[15] "Protein.Accessions" "Protein.Descriptions"
[17] "Number.of.Missed.Cleavages" "Charge"
[19] "Original.Precursor.Charge" "Delta.Score"
[21] "Delta.Cn" "Rank"
[23] "Search.Engine.Rank" "Concatenated.Rank"
[25] "mz.in.Da" "MHplus.in.Da"
[27] "Theo.MHplus.in.Da" "Delta.M.in.ppm"
[29] "Delta.mz.in.Da" "Ions.Matched"
[31] "Matched.Ions" "Total.Ions"
[33] "Intensity" "Activation.Type"
[35] "NCE.in.Percent" "MS.Order"
[37] "Isolation.Interference.in.Percent" "SPS.Mass.Matches.in.Percent"
[39] "Average.Reporter.SN" "Ion.Inject.Time.in.ms"
[41] "RT.in.min" "First.Scan"
[43] "Last.Scan" "Master.Scans"
[45] "Spectrum.File" "File.ID"
[47] "Abundance.126" "Abundance.127N"
[49] "Abundance.127C" "Abundance.128N"
[51] "Abundance.128C" "Abundance.129N"
[53] "Abundance.129C" "Abundance.130N"
[55] "Abundance.130C" "Abundance.131"
[57] "Quan.Info" "Peptides.Matched"
[59] "XCorr" "Number.of.Protein.Groups"
[61] "Contaminant" "Percolator.q.Value"
[63] "Percolator.PEP" "Percolator.SVMScore" We can see the quantitation data is located in columns 47 - 56 which start with “Abundance…” followed by the identifying TMT tag. Naming of the abundance columns is dependent on the third party software used and version, please make sure you check you identify the relevant column numbers containing the quantitation data in your own data.
We will pass this information to the quantCols argument when we use the readQFeatures function. As this is the first data import before any processing so let’s call this data level "psms_raw".
## Import data into QF object
cc_qf <- readQFeatures(assayData = df,
quantCols = 47:56,
name = "psms_raw")Checking arguments.Loading data as a 'SummarizedExperiment' object.Formatting sample annotations (colData).Formatting data as a 'QFeatures' object.
NoteFinding help files
During this course we see many new functions. For more information on any R function type ? and the name of the function in the R console to bring up the relevant documentation. Or go to the Help menu in RStudio and Help -> Search R Help. For example, to see the help files and documentation for the readQFeatures function type ?readQFeatures in your RStudio console. The help documentation highlights what structure the input data should be (a vector, dataframe or matrix etc.) as well as which arguments are required or optional. When using more complex functions, such as those which we will use for statistical analysis, it is advisable to consult the help documentation to ensure that you understand what the function’s default parameters are. This will help you to make sure that the function does what you are expecting it to do.
2.6 Accessing information within a QFeatures object
Let’s take a look at our newly created object.
cc_qfAn instance of class QFeatures (type: bulk) with 1 set:
[1] psms_raw: SummarizedExperiment with 45803 rows and 10 columns We see that we have created a QFeatures object containing a single SummarizedExperiment called “psms_raw”. There are 45803 rows in the data, representing 45803 PSMs. We also see that there are 10 columns representing our 10 samples.
We can access individual SummarizedExperiments within a QFeatures object using standard double bracket nomenclature (this is how you would normally access items of a list in R). As with all indexing we can use the list position or dataset name.
## Indexing using position
cc_qf[[1]]class: SummarizedExperiment
dim: 45803 10
metadata(0):
assays(1): ''
rownames(45803): 1 2 ... 45802 45803
rowData names(54): Checked Tags ... Percolator.PEP Percolator.SVMScore
colnames(10): Abundance.126 Abundance.127N ... Abundance.130C
Abundance.131
colData names(0):## Indexing using name
cc_qf[["psms_raw"]]class: SummarizedExperiment
dim: 45803 10
metadata(0):
assays(1): ''
rownames(45803): 1 2 ... 45802 45803
rowData names(54): Checked Tags ... Percolator.PEP Percolator.SVMScore
colnames(10): Abundance.126 Abundance.127N ... Abundance.130C
Abundance.131
colData names(0):Within each SummarizedExperiment(SE), the rowData, colData and assay data are accessed using rowData(), colData() and assay(), respectively. Let’s use these functions to explore the data structure further.
2.6.1 The assay container
Let’s start with the quantitation data of our SE called psms_raw. This is stored in the assay slot.
assay(cc_qf[["psms_raw"]])You will see that the R console is populated with the quantitation data and the whole dataset is too big to print to the screen. Let’s use the head command to show only the 6 lines (default) of the quantitation data.
Type into your R console,
head(assay(cc_qf[["psms_raw"]])) Abundance.126 Abundance.127N Abundance.127C Abundance.128N Abundance.128C
1 22.3 48.6 15.1 26.8 39.0
2 19.8 31.3 14.1 19.5 32.1
3 2.4 7.9 1.2 2.3 4.1
4 2.2 NA NA NA NA
5 2.2 4.2 2.2 1.6 5.7
6 NA 1.7 NA NA NA
Abundance.129N Abundance.129C Abundance.130N Abundance.130C Abundance.131
1 21.6 41.6 38.7 21.2 34.7
2 29.2 37.1 41.2 30.0 34.9
3 1.0 3.6 7.0 3.1 2.7
4 2.3 3.1 NA 1.7 NA
5 1.4 NA 3.6 4.0 4.0
6 NA NA NA NA 2.7This is equivalent to,
cc_qf[["psms_raw"]] %>%
assay() %>%
head() Abundance.126 Abundance.127N Abundance.127C Abundance.128N Abundance.128C
1 22.3 48.6 15.1 26.8 39.0
2 19.8 31.3 14.1 19.5 32.1
3 2.4 7.9 1.2 2.3 4.1
4 2.2 NA NA NA NA
5 2.2 4.2 2.2 1.6 5.7
6 NA 1.7 NA NA NA
Abundance.129N Abundance.129C Abundance.130N Abundance.130C Abundance.131
1 21.6 41.6 38.7 21.2 34.7
2 29.2 37.1 41.2 30.0 34.9
3 1.0 3.6 7.0 3.1 2.7
4 2.3 3.1 NA 1.7 NA
5 1.4 NA 3.6 4.0 4.0
6 NA NA NA NA 2.7The first code chunk uses nested functions and the second uses pipes. Throughout this course for ease of coding and clarity we will use pipes and where appropriate follow the tidyverse style of coding for clarity.
cc_qf[["psms_raw"]] %>%
assay() %>%
ncol()[1] 10We see the assay slot contains 10 quantitative columns corresponding to the 10 samples in our experiment. We see some quantitative values and some missing values, here denotated NA.
NoteThe dplyr pipe
In this course we will frequently use pipes, specifically the dplyr pipe part of the dplyr package and tidyverse. The pipe operator allows you to chain together multiple operations and code complex tasks in a more linear and understandable way, rather than having to nest multiple function calls or write multiple lines of code. For more information see Hadley Wickham’s Tidyverse at https://www.tidyverse.org.
2.6.2 The rowData container
Now let’s examine the the rowData slot,
cc_qf[["psms_raw"]] %>%
rowData() %>%
names() [1] "Checked" "Tags"
[3] "Confidence" "Identifying.Node.Type"
[5] "Identifying.Node" "Search.ID"
[7] "Identifying.Node.No" "PSM.Ambiguity"
[9] "Sequence" "Annotated.Sequence"
[11] "Modifications" "Number.of.Proteins"
[13] "Master.Protein.Accessions" "Master.Protein.Descriptions"
[15] "Protein.Accessions" "Protein.Descriptions"
[17] "Number.of.Missed.Cleavages" "Charge"
[19] "Original.Precursor.Charge" "Delta.Score"
[21] "Delta.Cn" "Rank"
[23] "Search.Engine.Rank" "Concatenated.Rank"
[25] "mz.in.Da" "MHplus.in.Da"
[27] "Theo.MHplus.in.Da" "Delta.M.in.ppm"
[29] "Delta.mz.in.Da" "Ions.Matched"
[31] "Matched.Ions" "Total.Ions"
[33] "Intensity" "Activation.Type"
[35] "NCE.in.Percent" "MS.Order"
[37] "Isolation.Interference.in.Percent" "SPS.Mass.Matches.in.Percent"
[39] "Average.Reporter.SN" "Ion.Inject.Time.in.ms"
[41] "RT.in.min" "First.Scan"
[43] "Last.Scan" "Master.Scans"
[45] "Spectrum.File" "File.ID"
[47] "Quan.Info" "Peptides.Matched"
[49] "XCorr" "Number.of.Protein.Groups"
[51] "Contaminant" "Percolator.q.Value"
[53] "Percolator.PEP" "Percolator.SVMScore" The columns of our rowData contain non-quantitative information derived from the third party identification search. The exact names of variables will differ between software and change over time, but the key information that we need to know about each feature (here PSMs) will always be here.
This information includes the sequence of the peptide to which each PSM corresponds ("Sequence") as well as the master protein to which the peptide is assigned ("Master.Protein.Accessions"). We will come across more of the variables stored in the rowData when we come to data cleaning and filtering.
We again use the head command to print the first 6 rows of the rowData
cc_qf[["psms_raw"]] %>%
rowData() %>%
head()DataFrame with 6 rows and 54 columns
Checked Tags Confidence Identifying.Node.Type Identifying.Node
<character> <logical> <character> <character> <character>
1 False NA High Sequest HT Sequest HT...
2 False NA High Sequest HT Sequest HT...
3 False NA High Sequest HT Sequest HT...
4 False NA High Sequest HT Sequest HT...
5 False NA High Sequest HT Sequest HT...
6 False NA High Sequest HT Sequest HT...
Search.ID Identifying.Node.No PSM.Ambiguity Sequence
<character> <integer> <character> <character>
1 A 2 Unambiguou... REEEMR
2 A 2 Unambiguou... QQNGTASSR
3 A 2 Unambiguou... CHMEENQR
4 A 2 Unambiguou... QACQER
5 A 2 Unambiguou... HSEAATAQR
6 A 2 Unambiguou... QQQQQQQHQQ...
Annotated.Sequence Modifications Number.of.Proteins Master.Protein.Accessions
<character> <character> <integer> <character>
1 rEEEmR N-Term(TMT... 2 P49755; Q1...
2 qQnGTASSR N-Term(TMT... 1 Q92917
3 cHmEENQR N-Term(TMT... 1 Q96TC7
4 qAcQER N-Term(TMT... 1 P26358
5 hSEAATAQR N-Term(TMT... 1 Q14103
6 qQQQQQQHQQ... N-Term(TMT... 1 Q6Y7W6
Master.Protein.Descriptions Protein.Accessions Protein.Descriptions
<character> <character> <character>
1 Transmembr... P49755; Q1... Transmembr...
2 G-patch do... Q92917 G-patch do...
3 Regulator ... Q96TC7 Regulator ...
4 DNA (cytos... P26358 DNA (cytos...
5 Heterogene... Q14103 Heterogene...
6 GRB10-inte... Q6Y7W6 GRB10-inte...
Number.of.Missed.Cleavages Charge Original.Precursor.Charge Delta.Score
<integer> <integer> <integer> <numeric>
1 1 2 2 0.1696
2 0 2 2 0.1154
3 0 3 3 1.0000
4 0 2 2 0.2849
5 0 2 2 0.4780
6 0 3 3 0.4098
Delta.Cn Rank Search.Engine.Rank Concatenated.Rank mz.in.Da
<numeric> <integer> <integer> <integer> <numeric>
1 0 1 1 3 547.776
2 0 1 1 1 589.802
3 0 1 1 1 450.202
4 0 1 1 1 510.758
5 0 1 1 1 600.320
6 0 1 1 1 635.659
MHplus.in.Da Theo.MHplus.in.Da Delta.M.in.ppm Delta.mz.in.Da Ions.Matched
<numeric> <numeric> <numeric> <numeric> <character>
1 1094.55 1094.55 -0.99 -0.00054 0/0
2 1178.60 1178.60 0.06 0.00003 0/0
3 1348.59 1348.59 -1.08 -0.00049 0/0
4 1020.51 1020.51 -1.44 -0.00074 0/0
5 1199.63 1199.63 -0.33 -0.00020 0/0
6 1904.96 1904.96 -1.33 -0.00084 0/0
Matched.Ions Total.Ions Intensity Activation.Type NCE.in.Percent MS.Order
<integer> <integer> <numeric> <character> <numeric> <character>
1 0 0 505446.3 CID NA MS2
2 0 0 197059.8 CID NA MS2
3 0 0 76685.6 CID NA MS2
4 0 0 103765.7 CID NA MS2
5 0 0 238514.8 CID NA MS2
6 0 0 76203.7 CID NA MS2
Isolation.Interference.in.Percent SPS.Mass.Matches.in.Percent
<numeric> <integer>
1 0.00000 50
2 47.04837 50
3 30.96584 50
4 3.10827 60
5 12.56510 70
6 0.00000 80
Average.Reporter.SN Ion.Inject.Time.in.ms RT.in.min First.Scan Last.Scan
<numeric> <numeric> <numeric> <integer> <integer>
1 30.8 60 39.4997 5816 5816
2 28.7 60 39.8252 5902 5902
3 3.5 60 40.0773 5967 5967
4 0.9 60 40.3077 6035 6035
5 2.8 60 40.7688 6151 6151
6 0.4 60 40.8788 6183 6183
Master.Scans Spectrum.File File.ID Quan.Info Peptides.Matched XCorr
<integer> <character> <character> <logical> <integer> <numeric>
1 5810 NocArrest_... F1.1 NA 104 1.71
2 5898 NocArrest_... F1.1 NA 125 1.82
3 5961 NocArrest_... F1.1 NA 4 0.94
4 6031 NocArrest_... F1.1 NA 105 1.86
5 6147 NocArrest_... F1.1 NA 136 1.82
6 6177 NocArrest_... F1.1 NA 285 1.22
Number.of.Protein.Groups Contaminant Percolator.q.Value Percolator.PEP
<integer> <character> <numeric> <numeric>
1 2 False 0.0033430 0.030030
2 1 False 0.0009278 0.006729
3 1 False 0.0014400 0.012330
4 1 False 0.0009278 0.006435
5 1 False 0.0004399 0.002259
6 1 False 0.0006928 0.003668
Percolator.SVMScore
<numeric>
1 0.132
2 0.352
3 0.260
4 0.360
5 0.536
6 0.4532.6.3 The colData container
The final slot within our SummarizedExperiment is the colData.
cc_qf[["psms_raw"]] %>%
colData()DataFrame with 10 rows and 0 columnsWe see this is also a DataFrame structure.
cc_qf[["psms_raw"]] %>%
colData() %>%
class()[1] "DFrame"
attr(,"package")
[1] "S4Vectors"It has 10 rows, one per sample, but no columns yet. This is because the sample metadata is not derived from an identification search and does not appear in our output .txt file. If we want to store extra information about our samples, we need to add this to the colData ourselves.
Each row of this 10 by 0 DataFrame corresponds to some feature of the quantitation channel. The names of these rows are the TMT sample names,
cc_qf[["psms_raw"]] %>%
colData() %>%
rownames() [1] "Abundance.126" "Abundance.127N" "Abundance.127C" "Abundance.128N"
[5] "Abundance.128C" "Abundance.129N" "Abundance.129C" "Abundance.130N"
[9] "Abundance.130C" "Abundance.131" 2.6.4 Annotating samples by adding meta-data
The colData contains information about the samples. We can create a data.frame in R with information regarding the samples, replicates, conditions and TMT tag used or we can read in this information into R from a spreadsheet or .csv.
Let’s open the file coldata.csv and then read it into R and add it to our QFeatures object. We use the read.csv function and pass the argument row.names = 1 which tells the function that the in the .csv file the row names are located in column 1.
## Read in coldata .csv
metadata_df <- read.csv("data/samples_meta_data.csv", row.names = 1)
## View data
metadata_df sample rep condition tag
Abundance.126 Control NA Pre-treatment 126
Abundance.127N M_1 1 M 127N
Abundance.127C M_2 2 M 127C
Abundance.128N M_3 3 M 128N
Abundance.128C G1_1 1 G1 128C
Abundance.129N G1_2 2 G1 129N
Abundance.129C G1_3 3 G1 129C
Abundance.130N DS_1 1 Desynch 130N
Abundance.130C DS_2 2 Desynch 130C
Abundance.131 DS_3 3 Desynch 131This .csv file has 10 rows, importantly the rows are annotated using the names of the rowData of our object, e.g. “Abundance.126”, “Abundance.127N” etc. These names must match exactly to the column names of the samples we have in our existing QFeatures object.
We can add information from this .csv to our colData
## Annotate colData with the sample data
colData(cc_qf) <- metadata_df
## Verify
colData(cc_qf)DataFrame with 10 rows and 4 columns
sample rep condition tag
<character> <integer> <character> <character>
Abundance.126 Control NA Pre-treatm... 126
Abundance.127N M_1 1 M 127N
Abundance.127C M_2 2 M 127C
Abundance.128N M_3 3 M 128N
Abundance.128C G1_1 1 G1 128C
Abundance.129N G1_2 2 G1 129N
Abundance.129C G1_3 3 G1 129C
Abundance.130N DS_1 1 Desynch 130N
Abundance.130C DS_2 2 Desynch 130C
Abundance.131 DS_3 3 Desynch 131## Apply this to the first set so that it is carried up
colData(cc_qf[["psms_raw"]]) <- colData(cc_qf)We can also change the names of our samples to simplify our downstream coding as well as our visualisation. This is done by changing the colnames.
## Change col names to represent the sample
colnames(cc_qf[["psms_raw"]]) <- cc_qf$sample
## Verify
colData(cc_qf[["psms_raw"]])DataFrame with 10 rows and 4 columns
sample rep condition tag
<character> <integer> <character> <character>
Control Control NA Pre-treatm... 126
M_1 M_1 1 M 127N
M_2 M_2 2 M 127C
M_3 M_3 3 M 128N
G1_1 G1_1 1 G1 128C
G1_2 G1_2 2 G1 129N
G1_3 G1_3 3 G1 129C
DS_1 DS_1 1 Desynch 130N
DS_2 DS_2 2 Desynch 130C
DS_3 DS_3 3 Desynch 131
TipKey Points
- The
QFeaturesinfrastructure provides a convenient and transparent way to store proteomics data across multiple levels. - A
QFeaturesobject stores each data level as aSummarizedExperiment, also called anset(dataset), with explicit links maintained between features across differentsets(e.g., links between all PSMs contributing to a peptide). - Explicit links between
setsare (i) generated automatically when aggregating data usingaggregateFeaturesor (ii) added manually using theaddAssayLinksfunction, as we will see later on. - Within each
SummarizedExperimentthe feature data is stored within therowData, quantitative data in theassay, and sample metadata in thecolData. These slots are accessible using their corresponding functions.